News
Updated chromosome-scale CH assembly and CH/CHO-K1 annotations
January 27, 2021
A significantly more continuous version of the Chinese hamster genome, CH PICRH (GCF_003668045.3), along with its 2020 RefSeq annotation is now on CHOgenome.org. Long-range scaffolding of the previous CH genome assembly was performed using high-throughput chromosome conformation capture (Hi-C) and now 97% of the genome is contained in 11 large scaffolds corresponding to the CH chromosomes. In addition, the updated 2020 RefSeq annotation for the CHO-K1 cell line is available. Both can be searched (gene search and BLAST) and viewed on JBrowse. More information on the most recent version of the Chinese hamster genome can be found here.
Updated CH assembly and CH/CHO-K1 annotations
May 1, 2019
We are excited to announce that the significantly improved Chinese hamster genome, CH PICR (GCF_00366804.1), along with its 2018 RefSeq annotation is now online. In addition, an updated RefSeq annotation for the CHO-K1 cell line is now available. Both can be searched (gene search and BLAST) and viewed on JBrowse. More information on the updated Chinese hamster genome can be found here.
New mRNA Expression Browser (Beta)
June 7, 2017
The browser (beta version) for the visualization of mRNA expression from CHO-K1 cells is now online. The data is from several published DNA-microarray or RNA-Seq experiments. The tutorial on how to use this browser can be found here.
Tutorial 5 – Viewing the CHO & CH Genomes
August 2014, Version 2.0
Click here for PDF version.
The genome viewer is a tool to provide visualization of the Chinese hamster (CH) and Chinese hamster ovary (CHO) cell genomes. This tool currently provides visualization via JBrowse of the 2014 annotated CHO-K1 genome and the chromosome-sorted CH genome.
CHO-K1 Genome Viewer
The genome viewer for the CHO-K1 cell line can be reached by selecting the "Genome Viewer" resource from the Resources tab or by selecting the "CHO-K1 Genome Viewer" button from the homepage.

CH Genome Viewer
The genome viewer for the chromosome-sorted Chinese hamster genome (Brinkrolf et al) can be reached by selecting the "CH Genome Viewer" button on the home page.

JBrowse
-
Loading Scaffolds
The 2014 CHO-K1 genome annotation is viewed using the JBrowse tool. The scaffold, not the chromosome, is the largest sequence assembly in this annotation. Scaffolds are labeled NW_0036#####.#, where # is equivalent to any digit from 0 to 9. If the desired scaffold ID number and range are known, they can be entered into the text boxes next to the zoom buttons (circled in green below). When the "Go" button is selected, the entered scaffold and range will be displayed. The scaffolds can also be selected from a drop down menu by clicking the down arrow next to the scaffold name. These scaffolds are sorted by size, with the longest scaffold at the top.
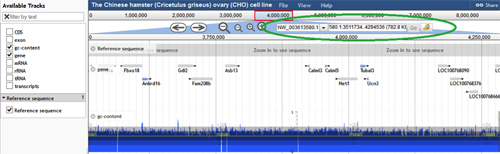
At the top of the graphic, the track called "Reference sequence" enables the user to view the region of choice. Once zoomed in far enough, this track displays the nucleotides on the scaffold. It shows the three possible translational frames for the nucleotide sequence and marks start (green) and stop (red) codons as well. The user is able to scroll the genome viewer tool using the arrows and zoom in using the (+/-) magnifying glasses above the "Reference Sequence" bar. The user can also zoom in to a specific area by double clicking on that area on the "Reference Sequence" track.
-
Feature Tracks
The tracks that are commonly filled on the gene viewer are GC Content, Gene, Exon, and mRNAs. The gc-content bar displays the percentage of bp's that are either a G or a C (calculated in a window of 100 bp), while the gene, exon, and mRNA tracks display the locations and descriptions of their respective genome features. By each track's label, there a (x) option which enables the user to turn off the track.
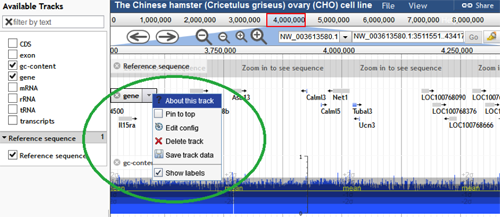
Hovering the mouse pointer over the track label, reveals a down arrow/menu with several options:
- "About this track" provides additional information about the specific track.
- "Pin to top" checkbox moves and keeps that track at the top of the track list when selected.
- "Edit config" enables the user to alter the settings of the track from the default values.
- "Delete track" permanently deletes the track for the current session.
- "Save track data" allows the user to download and save the track.
- "Show labels" checkbox makes the feature labels visible when checked. For instance, for the track gene, the gene names are visible when this box is checked.
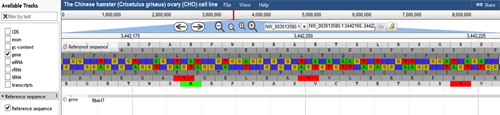
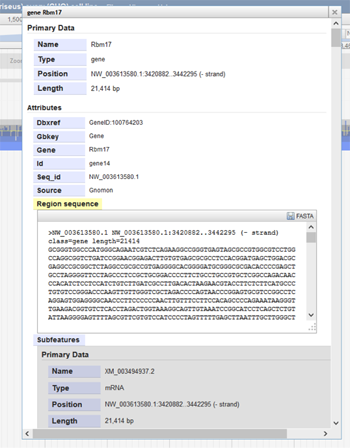
If the user clicks on a specific feature in a track, information about that feature will be provided. The image below shows information about the gene Rbm17. The pop-up window became visible when Rbm17 feature was clicked on in the gene track. Here, the gene's position, length, IDs, and nucleotide sequence are shown. Information about the gene's subfeatures, such as exons and mRNA, are also included. The FASTA sequence of the gene can be downloaded from this pop-up window by clicking on the "FASTA" save image at the top-right of the sequence.
-
Downloading Sequences
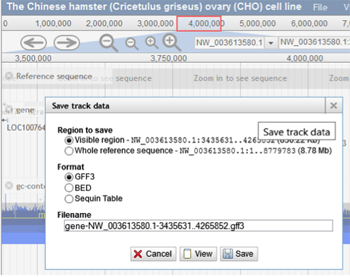
The "Save track data" option can be used to download the annotations for the visible region or the entire scaffold. The type of file can be selected from the list of file options, including GFF3, BED, and Sequin Table. The user can then specify the filename and view it before downloading. The reference sequence can also be downloaded as a FASTA file by choosing the "Save track data" option for the track called "Reference sequence".
-
Browser Configuration
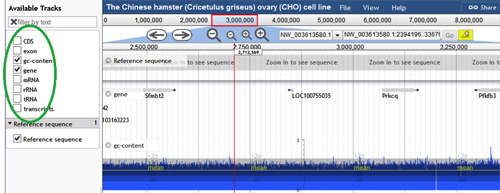
Add/Remove Tracks - To the left of the browser screen, there is a panel with a list of tracks. A track can be shown on the browser by checking the box next to the track name. Within the basic features, GC Content, Exon, Gene, and CDS are all preloaded tracks. Additional general tracks that are preloaded include mRNA, transcripts, rRNAs, and tRNAs. Any combination of these tracks may be enabled or disabled by the user.
Upload Custom Tracks – To upload a custom track, click on File>Open (file is located right to the title of the browser). The following box will appear:
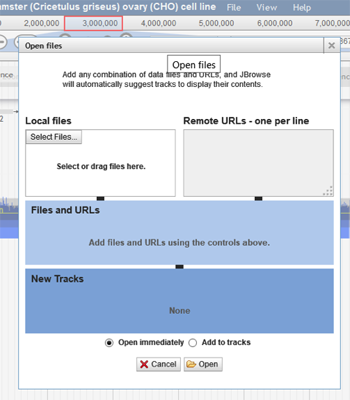
The user can select a file from their computer or use a remote URL to access the track. The track can be opened immediately on the browser or added to the tracks.
A user can also create a combination track by clicking File>Combination tracks. A new track is then added to the browser where the user can click and drag the tracks they want to combine. The following box appears:
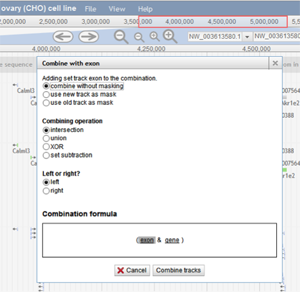
A user can choose how they want the tracks to be combined. In the image above, the intersection of the exon features and the gene features will be shown once the user chooses "Combine tracks".
Highlighting Regions –There are two ways a user can highlight a specified region on the browser. The first option is to go to the View menu (circled in green) and select "Set highlight". A text box appears where the user can fill in the genome coordinates they want highlighted. The default region specified is the complete region that is visible in the browser. The highlight can then be cleared using "Clear highlight". The third item on this menu, "Resize quant tracks", enables the user to change the height of the quantitative tracks.
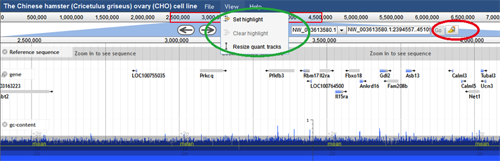
The second method to highlight regions is to click on the highlighter button (circled in red). The user can then select the region they want highlighted by clicking and dragging the mouse over the region directly on the browser.
-
CH Genome Viewer
The chromosomes from the Chinese hamster genome is also viewed using the JBrowse tool. The user must choose which chromosome they want to view first.

Once the chromosome is chosen, the user will be taken to the browser where they can view the reference sequence (no pre-loaded feature tracks are available). The instructions above apply to this genome viewer as well.
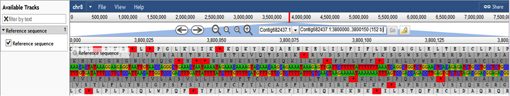
The image below shows ~150 bp of contig682437.1 from chromosome 8.